О возможной роли checkpoint-ингиборов в профилактике и лечении инфекционных заболеваний
В журнале «Nature Reviews Immunology» за 2017 год Michelle N. Wykes, Sharon R. Lewin была представлена очень интересная статья, посвященная вопросу возможного применения checkpoint-ингибиторов с целью профилактики и лечения целого ряда инфекционных заболеваний. В данной статье описывается значение передачи сheckpoint-сигналов в случае заражения малярией, ВИЧ и вирусом гепатита В, а также туберкулеза (ТБ), а также возможности терапевтического воздействия на иммунные контрольные точки в условиях наличия инфекционного заболевания.
При развитии острых инфекций, таких как малярия, а также при хронических вирусных инфекциях, включая ВИЧ и вирус гепатита, наблюдается повышение активности точек иммунного контроля - белка запрограммированной клеточной гибели 1 (PD1) и цитотоксического Т-лимфоцитарного антигена 4 (CTLA4).
Успешное применение checkpoint-ингибиторов в терапии злокачественных новообразований дает основание авторам статьи предполагать, что использование препаратов данной группы также будет эффективным для профилактики и лечения ряда социально-значимых инфекционных заболеваний.
Молекулы иммунной контрольной точки являются ингибиторными рецепторами, экспрессируемыми на иммунных клетках, которые являются триггером иммуносупрессивных сигнальных путей. Опосредованная передача сигналов через данные молекулы может привести к истощению иммунных клеток (особенно Т-лимфоцитов). Истощение Т-клеток проявляется снижением их функции, устойчивой экспрессией молекул иммунной контрольной точки (например, белка запрограммированной клеточной гибели 1 (PD1)), плохим обратным ответом и транскрипционным состоянием, отличным от состояния нормально функционирующих эффекторных Т-лимфоцитов или Т-клеток памяти[3]. Существует множество типов активирующих и ингибиторных взаимодействий, которые возникают между антиген-представляющими клетками (АРС) и Т-клетками, и эти взаимодействия определяют характер иммунных реакций.
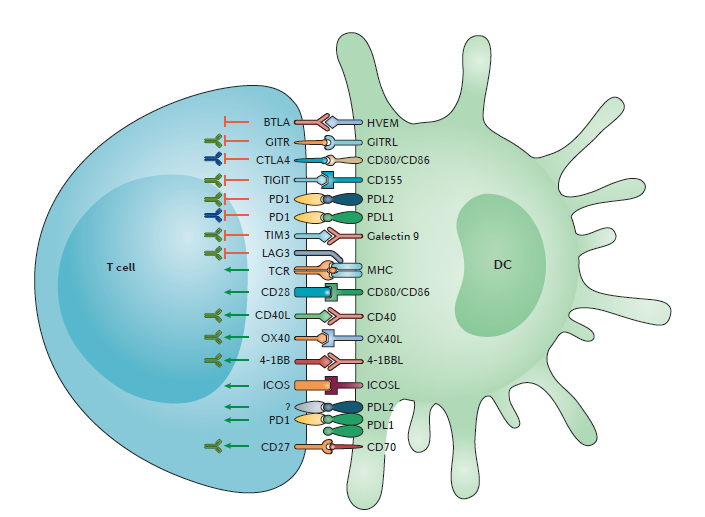
Рисунок 1 - Взаимодействие с антиген-представляющими клетками, регулирующими ответы Т-клеток (адаптировано Michelle N. Wykes et al., 2017).
Исследование иммуносупрессивных взаимодействий привело к клиническому развитию и внедрению новых методов лечения рака, которые увеличивают возможности использования специфических антител в качестве checkpoint-ингибиторов. Антитела, нацеленные на PD1 (Пембролизумаб, Ниволумаб), на цитотоксический Т-лимфоцитарный антиген 4 (CTLA4) (Ипилимумаб) и лиганд программированной гибели клеток 1 (PDL1) (Атезолизумаб, Авелумаб и Дурвалумаб), в настоящее время рекомендованы для монотерапии различных видов злокачественных опухолей. Кроме того, было показано, что комбинированная таргетная терапия, нацеленная на PD1 и CTLA4, является более эффективной, чем любая другая терапия меланомы[4], хотя также имеет токсическое воздействие на организм.
Основная проблема иммунотерапии заключается в том, чтобы понять, почему существуют различные ответы на лечение. На данный момент, идет поиск предиктивных «биомаркеров» благоприятного клинического ответа. Например, выявление экспрессии PDL1 на опухолевых клетках помогает в поиске тех пациентов, которые в наибольшей степени ответят на терапию, в которой используется блокаторы PD1 или PDL1[5]. Учитывая стоимость и токсичность использования checkpoint-ингибиторов, определение биомаркеров в настоящее время является главным приоритетом. Эффективность иммунотерапии в лечении инфекционных заболеваний изучена не так хорошо. Однако, предполагается, что она может быть использована для профилактики и лечения инфекционных заболеваний как в острой, так и в хронической фазах инфекции. Разработка вакцин для целого ряда инфекционных заболеваний, включая малярию, вирус гепатита B (HBV) и ВИЧ, может быть улучшена за счет использования checkpoint-ингибиторов. Учитывая, что резистентность к лекарственным средствам, применяемых для лечения малярии[10] и многих других инфекциях возрастает, что ВИЧ и HBV требуют лечения и контроля в течение всей жизни, рассматриваются новые возможные стратегии лечения этих инфекций. Кроме того, также требуется параллельный поиск биомаркеров, использование которых было бы возможно для выбора наиболее приемлемой терапии и установления временных рамок, когда применение иммунотерапии наиболее эффективно.
Таблица 1 - Сheckpoint-рецепторы
Checkpoint рецептор |
Тип эффекторных клеток |
Лиганд |
Примечание |
TIM3 |
TH1-клетки |
Galectin 9 на APCs |
Галектин 9 индуцирует внутриклеточный поток кальция, агрегацию и гибель клеток TH1 in vitro. Связь TIM3 и PD1 идентифицирует CD8 + Т-клетки у мышей с фенотипом истощения. |
LAG3 |
Природные, индуцированные тимусом и индуцированные, адаптивные периферические клетки Treg |
MHC II класса на APCs |
LAG3 улучшает функцию клеток Treg. LAG3 и PD1 обычно выражены на анергических или истощенных Т-клетках, а комбинированная блокада может вылечить большинство мышей установленных опухолей, которые в значительной степени устойчивы к лечению одиночных антител |
CD96 и TIGIT |
T-клетки и NK |
CD155 на DCs |
CD96 и TIGIT вызывают иммунодепрессию, конкурируя с CD226 для CD155. Слитый белок TIGIT-Fc ингибирует активацию Т-клеток, генерируя регуляторные DC. |
BTLA |
Т и В-клетки
|
HVEM, выраженный большинством гемопоэтических, эндотелиальных и эпителиальных клеток |
Лигирование TNFSF14 с помощью HVEM может быть стимулирующим, тогда как связывание BTLA-HVEM считается кo-ингибирующим. BTLA была связана с дисфункцией Т-клеток во время рака, а двойная блокада BTLA и PD1 явно улучшает противоопухолевый иммунитет |
TNFSF14 |
Интендативные адаптивные иммунные клетки, включая Т-клетки |
||
GITR |
Тreg клетки на высоких уровнях, обычные Т-клетки на низких уровнях,
|
GITRL на APCs |
GITR играет ключевую роль, поддерживая клетки CD4 + CD25 + Treg. GITRL в основном экспрессируется на APC, и показано, что антитела к GITR способствуют противоопухолевому ответу через потерю стабильности линии клеток Treg |
VISTA |
Гемопоэтические клетки |
Неизвестно |
Доклинические исследования с блокадой VISTA показывают многообещающее улучшение противоопухолевых ответов Т-клеток и улучшение выживаемости |
Белки иммунной контрольной точки при малярии
Малярия - это антропоноз, вызванный простейшими паразитами рода Plasmodium и передающийся человеку москитами рода Anopheles.
Большинство случаев малярии вызваны Plasmodium falciparum и Plasmodium vivax, а в 2015 году во всем мире насчитывалось 212 миллионов новых случаев малярии, из которых 429 000 человек были связаны только с P. Falciparum[11]. За последние 20 лет было разработано и клинически апробировано более 100 вакцин для борьбы с малярией. Большинство из них были специально разработаны для таргетного воздействия на паразитов в печеночной стадии или кровяной стадии развития. Данный метод был основан на индукции защитных антител и CD4 + Т-клеток, хотя несколько вакцин были разработаны для генерации CD8 + Т-клеточных ответов.
Лучший вариант вакцины, выявленной на сегодняшний день, является вакцина RTS, S / AS01E; однако, ее эффективность в первый год введения была только 43,6% , а к четвертому году использования снизилась до 16,8% [12]. Очевидно, что требуется поиск новой тактики лечения, нацеленной на механизмы иммунного воздействия на паразитов. В некоторых исследованиях было показано, что при малярии на стадии эритроцитарной шизогонии антитела играют ключевую роль в защите организма, о чем свидетельствует перенос сыворотки от иммунизированных взрослых детям[17]. Было установлено, что Т-хелперы1 (TH1) имеют ключевое значение для контроля над количеством паразитов в крови и, таким образом, для предотвращения развития инфекционного процесса [18, 19]. Антитела играют роль в уничтожении оставшихся паразитов [20]. Антитела и CD8+ Т-клетки принимают участие в формировании иммунитета, предотвращая повторное заражение [22]. Исследования также показали, что при малярии активируется апоптоз В-клеток [23] памяти из-за нарушения функций дендритных клеток (DC) [24]. Это может объяснить, почему применение вакцин не было успешным. И, вероятно, поэтому роль экспрессии PD1 в настоящее время расценивается как основной фактор в утрате иммунитета против малярии.
Малярия и Т-клеточное истощение
В ходе исследований в районах Мали и Кении, эндемичных по малярии, было обнаружено, что у людей, недавно инфицированных P. falciparum, повышена экспрессия PD1 на CD4+ [26, 27] и CD8+ T-клетках [27]. Аналогичным образом, повышение процента CD4+ T-клеток у людей с острыми фазами инфекции, вызванной P. vivax, P. falciparum имело место повышение экспрессии CTLA4, OX40, связанный с глюкокортикоидом TNFR-родственный белок и CD69 [28], что указывает на роль регуляторных T-клеток в подавлении иммунитета к малярии и показывает варианты потенциальных мишеней для управления контрольной точкой. Было показано, что в то время как PDL1, экспрессируемый на дендритных клетках, действительно ослабляет иммунные ответы против малярии, белок PDL2, экспрессируемый на тех же клетках, улучшает иммунные ответы, ингибируя взаимодействия PDL1-PD1 [9]. Примечательно, что во время острой фазы инфекционного процесса PD1, как было показано, обусловливал 95%-ную потерю числа и функциональной способности паразитоспецифических CD8+ Т-клеток, которые необходимы для борьбы с хроническим заболеванием [21]. Тем самым экспрессия белка PDL2 предохраняет от развития фатальных осложнений малярии (рис. 2).
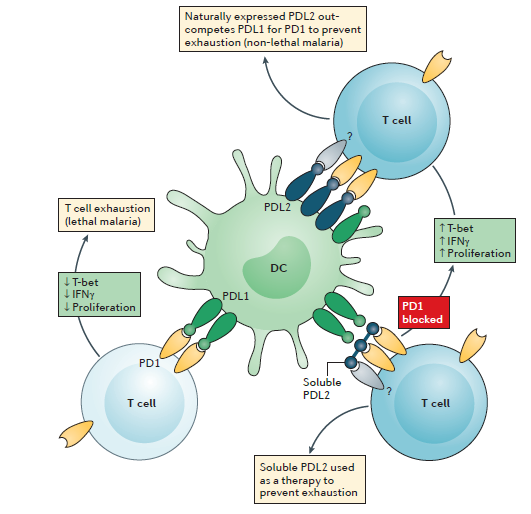
Рисунок 2 - PDL2 защищает от летальной малярии и обладает трансляционным потенциалом (адаптировано Michelle N. Wykes et al., 2017).
Применение checkpoint-ингибиторов при малярии
Было показано, что мультимерная форма PDL2, связанная с Fc-областью иммуноглобулина (PDL2-Fc) применима для нивелирования летальной инфекции и увеличивает шанс выживаемости после повторного инфицирования через несколько месяцев. Кроме того, комбинированная блокада ингибирующих молекул белка PDL1 и гена активации лимфоцитов 3 (LAG3) с антителами, ускорила выздоровление от острой нелетальной малярии в стадии эритроцитарной шизогонии, посредством улучшения функции CD4+клеток и увеличения титров антител [26]. Антитело-опосредованное инициирование передачи сигналов OX40 также увеличивало эффективность CD4+ T-хелперов, клеточного и гуморального иммунитета и, таким образом, улучшало эффективность уничтожения паразита при нелетальных формах малярийной инфекции [30].
Таким образом, отдельные белки контрольной точки способствуют развитию патогенеза малярии, и дальнейшее исследование их применения для терапевтических целей оправдано.
Эти методы лечения могут также иметь применение с целью «оживления» иммунных клеток, которые, как предполагается, не выполняют свою функцию у людей, проживающих в районах эндемичных по малярии [26,27]. Это даст возможность формировать стойкий иммунитет к малярии с помощью вакцин. Кроме того, использование checkpoint-ингибиторов может дополнять применение малярийных препаратов для выработки долгосрочного иммунитета, как это представлено на примере PDL2-Fc9.
Т-клеточное истощение и белки иммунной контрольной точки при ВИЧ-инфекции
Т-клеточное истощение является отличительной чертой многих хронических вирусных инфекций, включая ВИЧ. При отсутствии лечения ВИЧ-инфекции наблюдается повышенная экспрессия множественных белков иммунной контрольной точки, включая PD1, CTLA4, TIM3 и LAG3, как на CD4 +, так и на CD8 +клетках [36-38]. После АРТ экспрессия белков иммунной контрольной точки снижается, но остается повышенной по сравнению с контрольной группой, в которую входят люди, не имеющие ВИЧ-инфекции. Повышенная экспрессия PD1 преимущественно наблюдается в Т-клетках памяти, тогда как PD1 и CTLA4 экспрессируются регуляторными Т-клетками, а LAG3 экспрессируется в эффекторных Т-клетках памяти [41] (рис. 3).
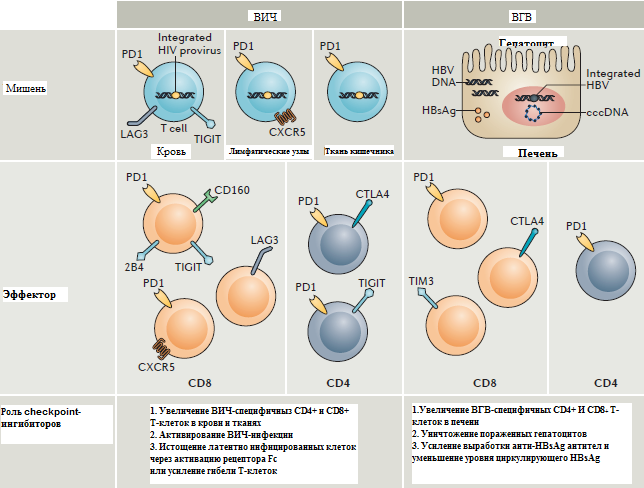
Рисунок 3 - Сheckpoints при ВИЧ-инфекции и инфекции вируса гепатита В (адаптировано Michelle N. Wykes et al., 2017).
Повышенные уровни экспрессии PD1 на общих и ВИЧ-специфических CD8+ Т-клетках при нелеченой ВИЧ-инфекции впервые были зарегистрированы более 10 лет назад.
В отсутствие AРТ увеличение экспрессии PD1 было связано с ускоренным снижением количества CD4+ Т-клеток в результате острой инфекции и нелеченой хронической инфекции. Многочисленные наблюдения продемонстрировали четкую связь между экспрессией PD1 на CD4+ или CD8+ Т-клетках и клиническим исходом.
Таблица 2 - Резюме доклинических или ex vivo исследований в области инфекционных заболеваний, сообщающих о преимуществах сheckpoint-ингибиторов
Инфекционное заболевание |
Тип аффекторных клеток |
Ингибиторные белки |
Виды мишеней |
Результаты |
ВИЧ |
CD4+ и CD8+ T клетки |
PD1, CTLA4, TIGIT и LAG3 |
Люди и мыши |
Увеличение ВИЧ-специфических CD8 + Т-клеток Проводятся клинические исследования при злокачественных новообразованиях |
ВГВ |
CD4+ и CD8+ T клетки |
PD1, CTLA4, 2B4 и TIM3 |
Люди (ex vivo), мыши и сурки |
PD1, CTLA4 и TIM3, экспрессируемые на CD4 + и CD8 + Т-клетках у пациентов с хронической инфекцией ВГВ PD1, CTLA4 и TIM3 улучшают функцию HBV-специфичных CD8 + Т-клеток in vitro |
ВГС |
CD4+ и CD8+ T клетки |
PD1 и PDL1 |
Люди |
Экспрессия PD1 повышается в отношении общего количества HCV-специфических CD8 + в периферической крови и печени пациентов с хронической инфекцией Блокада PDL1 восстанавливает функциональную компетентность CTL, специфичных для HCV, in vitro |
Туберкулез |
CD4+ и CD8+ T клетки |
TIM3 |
Мыши |
Блокада TIM3 восстанавливает функцию Т-клеток и улучшает бактериальный контроль, особенно у людей с хронической инфекцией |
Малярия |
CD4+ и CD8+ T клетки; B клетки |
PD1, PDL1, CTLA4, LAG3 и TIM3 |
Мыши |
Ускоренное уничтожение паразитов Выживание от летального заболевания Снижение заболеваемости церебральной малярией |
Использование сheckpoint-ингибиторов in vivo для SIV и ВИЧ-инфекции
По результатам исследований, введение антиPD1 антитела SIV-инфицированным макакам-резус привело к быстрому увеличению вирус-специфических CD8+ Т-клеток с более высоким функциональным качеством и снижению передачи сигналов интерферона, улучшение проницаемости кишечника [56].
Возможно, что эффективный ответ Т-клеток на анти-PD1-антитело требует наличия антигена, и что, поскольку АРТ приводит к резкому сокращению вирусных антигенов, функциональный ответ на блокаду иммунной контрольной точки может быть ограничен в этой ситуации.
Данные исследований свидетельствуют о том, что анти-CTLA4 антитела оказывают существенное влияние на ВИЧ, которое сохраняется при проведении АРТ. В основе этого лежит другой механизм действия анти-PD1-антитела, что приводит к снижению РНК ВИЧ в ткани лимфатических узлов. У людей, инфицированных ВИЧ, LAG3 также высоко экспрессируется на CD4+ и CD8+ Т-клетках в лимфатических узлах и крови, и это явление напрямую связано с уровнями РНК ВИЧ в плазме, но обратно пропорционально количеству лимфоцитов CD4+ [59].
Блокада TIGIT и PD1 с анти-TIGIT и anti-PDL1 антителами ex vivo привела к значительному улучшению ВИЧ-специфической функции CD4+ T-клеток у людей, инфицированных ВИЧ, в том числе и на АРT [38].
Белки иммунной контрольной точки и устойчивость ВИЧ
В отличие от злокачественных клеток, которые обычно экспрессируют лиганды белков иммунной контрольной точки, таких как PDL1 [62], у людей, инфицированных ВИЧ, в том числе и при проведении АРТ, сами белки иммунной контрольной точки избирательно распознают клетки, инфицированные ВИЧ, которые сохраняются на фоне АРТ [63-64] (рис. 4).
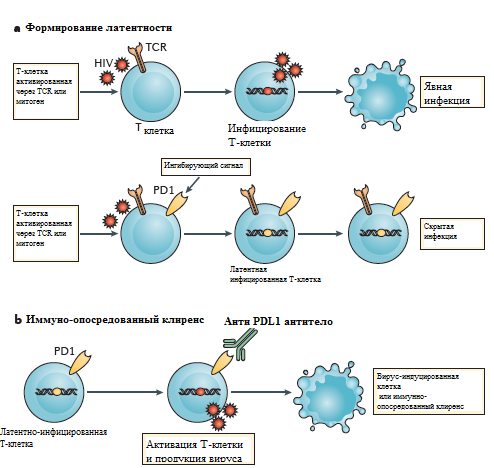
Рисунок 4 - Предполагаемая роль PD1 в формировании и изменении латентности ВИЧ (адаптировано Michelle N. Wykes et al., 2017).
Это наблюдение имеет большое значение для разработки метода по ликвидации резидуального вируса, который сохраняется, несмотря на проводимую АРТ, поскольку наличие инфицированных клеткок является основным препятствием для излечения. Многие исследования показали значительную корреляцию между частотой встречаемости PD1+ CD4+ T-клеток и PD1+ CD8+ T-клеток с различными маркерами устойчивости ВИЧ-инфекции к АРТ в крови [63,65,66], лимфатических узлах [67] и желудочно-кишечном тракте. Белки иммунной контрольной точки, отличные от PD1, также могут распознавать инфицированные клетки у лиц, получающих АРТ. В недавнем времени было показано, что ВИЧ может быть значительно обогащен клетками, полученными от людей, инфицированных ВИЧ и получающих AРT, которые экспрессировали PD1, TIGIT и LAG3 по сравнению с клетками, которые не экспрессировали ни один из этих белков [64](рис. 3). Внедрение CTLA4, опосредованное вирусным белком Nef, потенциально может играть роль в сохранении устойчивости ВИЧ в этих клетках [70]. Также было показано, что у человека с диагнозом «метастатическая меланома», инфицированного ВИЧ и получающего АРТ, наблюдается значительное увеличение связанной с клетками ВИЧ-РНК после лечения анти- CTLA4 антителами (Ипилимумаб) [71] и анти-PD1 антителами (Ниволумаб) [72]. Эти данные еще должны быть подтверждены в клинических исследованиях.
Использование сheckpoint-ингибиторов в качестве стратегии лечения ВИЧ
Изучение доза-зависимой фазы анти-PDL1-терапии было прекращено после введения самой низкой дозы 6 людям с ВИЧ-инфекцией получающим АРТ [73], в связи с развитием токсического воздействия на сетчатку. На сегодняшний день лишь немногие люди с ВИЧ-инфекцией получили зарегистрированные препараты сheckpoint-ингибиторов, поскольку лица с ВИЧ-инфекцией были исключены из исследования. Но в настоящее время использование данных препаратов для ВИЧ-инфицированных людей становится возможным.
Таким образом, сheckpoint-ингибиторы могут внести существенный вклад в достижение полного излечения от ВИЧ-инфекции и позволять людям безопасно прекращать АРТ.
Т-клеточное истощение и белки иммунных контрольных точек при HBV-инфекции
При хронической инфекции HBV, не подвергавшейся лечению, общие и HBV-специфичные CD8 + Т-клетки экспрессируют высокие уровни PD1, CTLA4 и TIM3 [84-86], а при острой инфекции HBV циркулирующие и внутрипеченочные CD8+ Т-клетки экспрессируют высокие уровни PD1 [87].
Было показано, что наличие лиганда для PD1, PDL1 более высокий на циркулирующих CD14+ моноцитах и CD19+ B-клетках у лиц с хронической инфекцией HBV, циррозом печени и HCC и, следовательно, может способствовать постоянному Т-клеточному истощению [88]. Исследования показали, что дефекты функции Т-клеток при хронической инфекции HBV не ограничиваются увеличением экспрессии белков иммунной контрольной точки. Внутрипеченочные Т-клетки также проявляют повышенную экспрессию PD1 [87] и TIM3 [84]. Галектин (лиганд TIM3) также был активен на клетках Купфера, что, возможно, позволяет поддерживать внутрипеченочное Т-клеточное истощение [84]. Внутрипеченочные CD8+ Т-клетки также экспрессируют другие белки истощения, включая BTLA, и могут продуцировать IL-10, что дополнительно ингибирует функцию Т-клеток [92].
Применение сheckpoint-ингибиторов в качестве стратегии лечения вирусного гепатита В
Исследования с использованием крови людей, страдающих с хроническим гепатитом В, продемонстрировали, что ингибирование PD1, CTLA4, 2B4 и TIM3 приводит к увеличению функции HBV-специфичных CD8+ Т-клеток [83,84,86,93–96]. Применение сheckpoint-ингибиторов в качестве как монотерапии, так и в сочетании с антивирусными препаратами, может увеличить продукцию HBV-специфичных CD8+ Т-клеток. А также влиять на выработку антител против HBsAg. Но при этом существуют значительные риски, в том числе повышенная инфильтрация ткани печени повторно активированными Т-клеткам, что может вызвать воспаление.
Установлено, что добавление анти-PDL1-антитела к вакцине и энтекавиру по сравнению с одной только вакциной и энтекавиром привело к значительному увеличению иммунологического и клинического ответа без развития гепатотоксичности [97]. Недавнее исследование open-label Ниволумаба (анти-PD1-Ат) с вакциной против HBV и без нее и 20 участниками с подавленной вирусной хронической инфекцией HBV показало, что Ниволумаб безопасен и хорошо переносится, а один участник подвергся сероконверсии HBsAg [99].
В настоящее время ведется множество клинических испытаний сheckpoint-ингибиторов у лиц с хроническим HBV. Таким образом, применение сheckpoint-ингибиторов играет важную роль в лечении вирусного гепатита В, ограничивая повреждение печени при острой инфекции и способствуя стабилизации процесса при хроническом HBV.
Белки иммунной контрольной точки при туберкулезе
Для формирования невосприимчивости организма к M. tuberculosis необходимы CD4+ Т-клетки. Было обнаружено, что TBC-специфические CD4+ Т-клетки у лиц с активной туберкулезной инфекцией продуцируют IFNγ, IL2 и TNF и экспрессируют PD1. Было показано, что функционально истощенные ТМ3 + Т-клетки накапливаются при наличии хронической туберкулезной инфекции. Ингибирование TIM3 восстанавливает функции Т-клеток и улучшает контроль за бактериальной нагрузкой у лиц с хроническим течением туберкулеза. Комбинация препаратов, рутинно используемых для лечения туберкулеза, с сheckpoint-ингибиторами даст возможность формирования иммунитета развиваться, при этом, бактериальная нагрузка будет находиться под частичным контролем.
Заключение
Лекарственная терапия малярии, ВИЧ-инфекции, вирусного гепатита В и туберкулеза в настоящее время является большой проблемой в связи с развитием полирезистентности микроорганизмов, при этом разработка эффективной вакцины зачастую невозможна. Поэтому применение сheckpoint-ингибиторов может оказаться абсолютно новой стратегией борьбы с хроническими инфекциями, для которых по-прежнему отсутствуют эффективные методы лечения, и возможности применения вакцин ограничены. Однако следует учесть, что применение данных препаратов в свою очередь имеет ограничения и побочные эффекты. В ряде исследований было выдвинуто предположение, что ингибирование иммунной контрольной точки также может быть использовано для лечения ряда инфекционных заболеваний, в том числе в терапии малярии, ВИЧ-инфекции, что несомненно требует подтверждения в клинических исследованиях.
Источники:
- Baumeister, S. H., Freeman, G. J., Dranoff, G. & Sharpe, A. H. Coinhibitory pathways in immunotherapy for cancer. Annu. Rev. Immunol. 34, 539–573 (2016).
- Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
- Wherry, E. J. T cell exhaustion. Nat. Immunol. 12, 492–499 (2011).
- Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
- Brahmer, J. R. et al. Phase I study of single-agent anti-programmed death?1 (MDX?1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 28, 3167–3175 (2010).
- Chen, P. L. et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 6, 827–837 (2016).
- Kamphorst, A. O. et al. Proliferation of PD?1+ CD8 T cells in peripheral blood after PD?1?targeted therapy in lung cancer patients. Proc. Natl Acad. Sci. USA 114, 4993–4998 (2017).
- Huang, A. C. et al. T?Cell invigoration to tumour burden ratio associated with anti?PD?1 response. Nature 545, 60–65 (2017).
- Karunarathne, D. S. et al. Programmed death?1 ligand 2?mediated regulation of the PD?L1 to PD?1 axis is essential for establishing CD4+ T cell immunity. Immunity 45, 333–345 (2016).
- An important study on the mechanism of the protective function of PDL2.
- Cheng, Q., Kyle, D. E. & Gatton, M. L. Artemisinin resistance in Plasmodium falciparum: a process linked to dormancy? Int. J. Parasitol. Drugs Drug Resist. 2, 249–255 (2012).
- World Health Organization. World Malaria Report (WHO, 2016).
- Olotu, A. et al. Four-year efficacy of RTS,S/AS01E and its interaction with malaria exposure. N. Engl. J. Med. 368, 1111–1120 (2013).
- Barragan, A., Kremsner, P. G., Weiss, W., Wahlgren, M. & Carlson, J. Age-related buildup of humoral immunity against epitopes for rosette formation and agglutination in African areas of malaria endemicity. Infect. Immun. 66, 4783–4787 (1998).
- Bull, P. C. et al. Parasite antigens on the infected red cell surface are targets for naturally acquired immunity to malaria. Nat. Med. 4, 358–360 (1998).
- Soe, S. et al. Premunition against Plasmodium falciparum in a malaria hyperendemic village in Myanmar. Trans. R. Soc. Trop. Med. Hyg. 95, 81–84 (2001).
- Mazier, D. et al. Hepatic phase of malaria is the target of cellular mechanisms induced by the previous and the subsequent stages. A crucial role for liver nonparenchymal cells. Immunol. Lett. 25, 65–70 (1990).
- Cohen, S., McGregor, I. & Carrington, S. Gamma-globulin and acquired immunity to human malaria. Nature 192, 733–737 (1961).
- Langhorne, J., Simon-Haarhaus, B. & Meding, S. J. The role of CD4+ T cells in the protective immune response to Plasmodium chabaudi in vivo. Immunol. Lett. 25, 101–107 (1990).
- Podoba, J. E. & Stevenson, M. M. CD4+ and CD8+ T lymphocytes both contribute to acquired immunity to blood-stage Plasmodium chabaudi AS. Infect. Immun. 59, 51–58 (1991).
- von der Weid, T., Honarvar, N. & Langhorne, J. Gene-targeted mice lacking B cells are unable to eliminate a blood stage malaria infection. J. Immunol. 156, 2510–2516 (1996).
- Horne-Debets, J. M. et al. PD?1 dependent exhaustion of CD8+ T cells drives chronic malaria. Cell Rep. 5, 1204–1213 (2013).
- The first study to show a role for CD8+ T cells in controlling blood-stage malaria.
- Horne-Debets, J. M. et al. Mice lacking Programmed cell death?1 show a role for CD8+ T cells in long-term immunity against blood-stage malaria. Sci. Rep. 6, 26210 (2016).
- Wykes, M. N., Zhou, Y. H., Liu, X. Q. & Good, M. F. Plasmodium yoelii can ablate vaccine-induced long-term protection in mice. J. Immunol. 175, 2510–2516 (2005).
- Liu, X. Q. et al. Malaria infection alters the expression of B?cell activating factor resulting in diminished memory antibody responses and survival. Eur. J. Immunol. 42, 3291–3301 (2012).
- Pierce, S. K. & Miller, L. H. World Malaria Day 2009: what malaria knows about the immune system that immunologists still do not. J. Immunol. 182, 5171–5177 (2009).
- Butler, N. S. et al. Therapeutic blockade of PD?L1 and LAG?3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 13, 188–195 (2012).
- The first study to show that checkpoint blockade during malaria could improve protective immunity.
- Illingworth, J. et al. Chronic exposure to Plasmodium falciparum is associated with phenotypic evidence of B and T cell exhaustion. J. Immunol. 190, 1038–1047 (2013).
- Goncalves-Lopes, R. M. et al. Surface expression of inhibitory (CTLA?4) and stimulatory (OX40) receptors by CD4+ regulatory T cell subsets circulating in human malaria. Microbes Infect. 18, 639–648 (2016).
- Hou, N. et al. T?Cell immunoglobulin- and mucin-domain-containing molecule 3 signaling blockade improves cell-mediated immunity against malaria. J. Infect. Dis. 214, 1547–1556 (2016).
- Zander, R. A. et al. PD?1 co?inhibitory and OX40 co?stimulatory crosstalk regulates helper T cell differentiation and anti-plasmodium humoral immunity. Cell Host Microbe 17, 628–641 (2015).
- Hafalla, J. C. et al. The CTLA?4 and PD?1/PD?L1 inhibitory pathways independently regulate host resistance to plasmodium-induced acute immune pathology. PLoS Pathog. 8, e1002504 (2012).
- Hisaeda, H. et al. Resistance of regulatory T cells to glucocorticoid-induced TNFR family-related protein (GITR) during Plasmodium yoelii infection. Eur. J. Immunol. 35, 3516–3524 (2005).
- Lepenies, B. et al. Ligation of B and T lymphocyte attenuator prevents the genesis of experimental cerebral malaria. J. Immunol. 179, 4093–4100 (2007).
- GBD 2015 HIV Collaborators et al. Estimates of global, regional, and national incidence, prevalence, and mortality of HIV, 1980–2015: the Global Burden of Disease Study 2015. Lancet HIV 3, e361–e387 (2016).
- Deeks, S. G. et al. International AIDS Society global scientific strategy: towards an HIV cure 2016. Nat. Med. 22, 839–850 (2016).
- Trautmann, L. et al. Upregulation of PD?1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 12, 1198–1202 (2006).
- One of the first papers to identify PD1 as a marker of T cell exhaustion in HIV infection.
- Kaufmann, D. E. et al. Upregulation of CTLA?4 by HIV-specific CD4+ T cells correlates with disease progression and defines a reversible immune dysfunction. Nat. Immunol. 8, 1246–1254 (2007).
- Chew, G. M. et al. TIGIT marks exhausted T cells, correlates with disease progression, and serves as a target for immune restoration in HIV and SIV Infection. PLoS Pathog. 12, e1005349 (2016).
- An examination of TIGIT and PD1 in T cell exhaustion in HIV infection both on and off ART.
- Rutishauser, R. et al. Early and delayed antiretroviral therapy (ART) result in comparable reductions in CD8+ T cell exhaustion marker expression. AIDS Res Hum Retroviruses 33, 658–667 (2017).
- Zhang, J. Y. et al. PD?1 up?regulation is correlated with HIV-specific memory CD8+ T?cell exhaustion in typical progressors but not in long-term nonprogressors. Blood 109, 4671–4678 (2007).
- Tian, X. et al. The upregulation of LAG?3 on T cells defines a subpopulation with functional exhaustion and correlates with disease progression in HIV-infected subjects. J. Immunol. 194, 3873–3882 (2015).
- Sauce, D. et al. PD?1 expression on human CD8 T cells depends on both state of differentiation and activation status. AIDS 21, 2005–2013 (2007).
- Day, C. L. et al. PD?1 expression on HIV-specific T cells is associated with T?cell exhaustion and disease progression. Nature 443, 350–354 (2006).
- Petrovas, C. et al. PD?1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 203, 2281–2292 (2006).
- Leong, Y. A. et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 17, 1187–1196 (2016).
- Mylvaganam, G. H. et al. Dynamics of SIV-specific CXCR5+ CD8 T cells during chronic SIV infection. Proc. Natl Acad. Sci. USA 114, 1976–1981 (2017).
- Li, S. et al. Simian immunodeficiency virus-producing cells in follicles are partially suppressed by CD8+ cells in vivo. J. Virol. 90, 11168–11180 (2016).
- Hoffmann, M. et al. Exhaustion of activated CD8 T cells predicts disease progression in primary HIV?1 Infection. PLoS Pathog. 12, e1005661 (2016).
- Shive, C. L. et al. Inflammation perturbs the IL?7 axis, promoting senescence and exhaustion that broadly characterize immune failure in treated HIV infection. J. Acquir. Immune Def. Syndr. 71, 483–492 (2016).
- Sinha, A. et al. role of T?cell dysfunction, inflammation, and coagulation in microvascular disease in HIV. J. Am. Heart Assoc. 5, e004243 (2016).
- Kelesidis, T. et al. Oxidized lipoproteins are associated with markers of inflammation and immune activation in HIV?1 infection. AIDS 30, 2625–2633 (2016).
- Hurst, J. et al. Immunological biomarkers predict HIV?1 viral rebound after treatment interruption. Nat. Commun. 6, 8495 (2015).
- The first publication to show a functional link between expression of immune checkpoint proteins on T cells prior to ART and the time to rebound after cessation of ART. An important observation in determining the role of immune checkpoint proteins and HIV cure or remission.
- Akhmetzyanova, I. et al. PD?L1 expression on retrovirus-infected cells mediates immune escape from CD8+ T cell killing. PLoS Pathog. 11, e1005224 (2015).
- Velu, V. et al. Enhancing SIV-specific immunity in vivo by PD?1 blockade. Nature 458, 206–210 (2009).
- Dyavar Shetty, R. et al. PD?1 blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J. Clin. Invest. 122, 1712–1716 (2012).
- Mylvaganam, G. H. et al. PD?1 blockade synergizes with ART for restoring anti-viral CD8 T cell function and possibly destabilizing the viral reservoir in SIV infected macaques [Abstract 9016]. Presented at the International AIDS Conference, Durban, South Africa, July 2016.
- Gill, A. L. et al. Programed death?1/programed death-ligand 1 expression in lymph nodes of HIV infected patients: results of a pilot safety study in rhesus macaques using anti-programed death-ligand 1 (Avelumab). AIDS 30, 2487–2493 (2016).
- Cecchinato, V. et al. Immune activation driven by CTLA?4 blockade augments viral replication at mucosal sites in simian immunodeficiency virus infection. J. Immunol. 180, 5439–5447 (2008).
- Hryniewicz, A. et al. CTLA?4 blockade decreases TGF- β, IDO, and viral RNA expression in tissues of SIVmac251?infected macaques. Blood 108, 3834–3842 (2006).
- Tauriainen, J. et al. Perturbed CD8+ T cell TIGIT/ CD226/PVR axis despite early initiation of antiretroviral treatment in HIV infected individuals. Sci. Rep. 7, 40354 (2017).
- Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag?3, Tim?3, and TIGIT: co?inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016).
- Chatterjee, S. et al. A humanized antibody for imaging immune checkpoint ligand PD?L1 expression in tumors. Oncotarget 7, 10215–10227 (2016).
- Chomont, N. et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15, 893–900 (2009). The first demonstration of the relationship between PD1 and HIV persistence in patients on ART.
- Fromentin, R. et al. CD4+ T cells expressing PD?1, TIGIT and LAG?3 contribute to HIV persistence during ART. PLoS Pathog. 12, e1005761 (2016). An important paper demonstrating that multiple immune checkpoint markers, not just PD1, are involved in HIV persistence in patients on ART.
- Hatano, H. et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells.. J. Infect. Dis. 208, 50–56 (2013).
- Cockerham, L. R. et al. Programmed death?1 expression on CD4+ and CD8+ T cells in treated and untreated HIV disease. AIDS 28, 1749–1758 (2014).
- Banga, R. et al. PD?1+ and follicular helper T cells are responsible for persistent HIV?1 transcription in treated aviremic individuals. Nat. Med. 22, 754–761 (2016).
- The first demonstration that TFH cells that express high levels of PD1 are an important reservoir for HIV in patients on ART.
- Khoury, G. et al. HIV persistence and T?cell activation in blood, rectal and lymph node tissue in HIV-infected individuals receiving suppressive ART. J. Infect. Dis. 215, 911–919 (2017).
- El?Far, M. et al. Nef promotes evasion of human immunodeficiency virus type 1?infected cells from the CTLA?4?mediated inhibition of T?cell activation. J. Gen. Virol. 96, 1463–1477 (2015).
- El?Far, M. et al. Down-regulation of CTLA?4 by HIV?1 Nef protein. PLoS ONE 8, e54295 (2013).
- Wightman, F. et al. Effect of ipilimumab on the HIV reservoir in an HIV-infected individual with metastatic melanoma. AIDS 29, 504–506 (2015).
- Van der Sluis, R. M. et al. Anti?PD?1 disrupts HIV latency in non-proliferating but not in proliferating T?cells [Abstract OA3?3]. J. Virus Eradication 3, Suppl. 1 (2017).
- Gay, C. L. et al. Clinical trial of the anti?PD?L1 antibody BMS?936559 in HIV?1 infected participants on suppressive antiretroviral therapy. J. Infect. Dis. 215, 1725–1733 (2017).
- Rasmussen, T. A., Anderson, J. L., Wightman, F. & Lewin, S. R. Cancer therapies in HIV cure research. Curr. Opin. HIV AIDS 12, 96–104 (2017).
- Romano, E. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl Acad. Sci. USA 112, 6140–6145 (2015).
- Dahan, R. et al. FcγRs modulate the anti-tumor activity of antibodies targeting the PD?1/PD?L1 axis. Cancer Cell 28, 285–295 (2015).
- An interesting paper exploring the future direction of immune checkpoint blockers with modifications to the Fc tail so that the antibody activates Fc receptors.
- Zhou, J. et al. PD1?based DNA vaccine amplifies HIV?1 GAG-specific CD8+ T cells in mice. J. Clin. Invest. 123, 2629–2642 (2013).
- Schweitzer, A., Horn, J., Mikolajczyk, R. T., Krause, G. & Ott, J. J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet 386, 1546–1555 (2015).
- Stanaway, J. D. et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. Lancet 388, 1081–1088 (2016).
- Revill, P., Testoni, B., Locarnini, S. & Zoulim, F. Global strategies are required to cure and eliminate HBV infection. Nat. Rev. Gastroenterol. Hepatol. 13, 239–248 (2016).
- Guidotti, L. G., Isogawa, M. & Chisari, F. V. Host-virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 36, 61–66 (2015).
- Chang, J. J. et al. The phenotype of hepatitis B virus-specific T cells differ in the liver and blood in chronic hepatitis B virus infection. Hepatology 46, 1332–1340 (2007).
- Boni, C. et al. Characterization of hepatitis B virus (HBV)-specific T?cell dysfunction in chronic HBV infection. J. Virol. 81, 4215–4225 (2007).
- Nebbia, G. et al. Upregulation of the Tim?3/galectin?9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE 7, e47648 (2012).
- Bengsch, B., Martin, B. & Thimme, R. Restoration of HBV-specific CD8+ T cell function by PD?1 blockade in inactive carrier patients is linked to T cell differentiation. J. Hepatol. 61, 1212–1219 (2014).
- Schurich, A. et al. Role of the coinhibitory receptor cytotoxic T lymphocyte antigen?4 on apoptosis-prone CD8 T cells in persistent hepatitis B virus infection. Hepatology 53, 1494–1503 (2011).
- Zhang, Z. et al. Dynamic programmed death 1 expression by virus-specific CD8 T cells correlates with the outcome of acute hepatitis B. Gastroenterology 134, 1938–1949 (2008).
- A demonstration of the importance of PD1 expression in acute HBV infection.
- Huang, Z. Y. et al. Clinical significance of dynamics of programmed death ligand?1 expression on circulating CD14+ monocytes and CD19+ B Cells with the progression of Hepatitis B virus infection. Viral Immunol. 30, 224–231 (2017).
- Raziorrouh, B. et al. Inhibitory phenotype of HBV-specific CD4+ T?cells is characterized by high PD?1 expression but absent coregulation of multiple inhibitory molecules. PLoS ONE 9, e105703 (2014).
- Fisicaro, P. et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 23, 327–336 (2017).
- Maini, M. K. et al. The role of virus-specific CD8+ cells in liver damage and viral control during persistent hepatitis B virus infection. J. Exp. Med. 191, 1269–1280 (2000).
- Wang, H. et al. Hepatic expansion of virus-specific CD8+BTLA+ T cells with regulatory properties in chronic hepatitis B virus infection. Cell. Immunol. 311, 36–45 (2017).
- Fisicaro, P. et al. Antiviral intrahepatic T?cell responses can be restored by blocking programmed death?1 pathway in chronic hepatitis B. Gastroenterology 138, 682–693 (2010).
- Raziorrouh, B. et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus-specific CD8+ T?cell function. Hepatology 52, 1934–1947 (2010).
- Isogawa, M., Furuichi, Y. & Chisari, F. V. Oscillating CD8+ T cell effector functions after antigen recognition in the liver. Immunity 23, 53–63 (2005).
- Maier, H., Isogawa, M., Freeman, G. J. & Chisari, F. V. PD?1:PD?L1 interactions contribute to the functional suppression of virus-specific CD8+ T lymphocytes in the liver. J. Immunol. 178, 2714–2720 (2007).
- Zhang, E. et al. The expression of PD?1 ligands and their involvement in regulation of T cell functions in acute and chronic woodchuck hepatitis virus infection. PLoS ONE 6, e26196 (2011).
- Liu, J. et al. Enhancing virus-specific immunity in vivo by combining therapeutic vaccination and PD?L1 blockade in chronic hepadnaviral infection. PLoS Pathog. 10, e1003856 (2014).
- A study showing the effect of anti?PDL1 antibody in an animal model of HBV; it reduced viral rebound after cessation of antiviral therapy and increased antibodies to HBV surface antigen.
- Gane, E. J. et al. A phase 1 study evaluating anti?PD?1 treatment with or without GS?4774 in HBeAg negative chronic hepatitis B patients [Abstract]. Eur. Associ. Study Liver Dis. (2017).
- El-Khoueiry, A. B. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet http://dx.doi.org/10.1016/ S0140-6736(17)31046-2 (2017).
- Pollock, K. M. et al. PD?1 expression and cytokine secretion profiles of Mycobacterium tuberculosis-specific CD4+ T?cell subsets; potential correlates of containment in HIV?TB co?Infection. PLoS ONE 11, e0146905 (2016).
- Boer, M. C. et al. KLRG1 and PD?1 expression are increased on T?cells following tuberculosis-treatment and identify cells with different proliferative capacities in BCG-vaccinated adults. Tuberculosis (Edinb). 97, 163–171 (2016)
- Barber, D. L., Mayer-Barber, K. D., Feng, C. G., Sharpe, A. H. & Sher, A. CD4 T cells promote rather than control tuberculosis in the absence of PD?1?mediated inhibition. J. Immunol. 186, 1598–1607 (2011).
- Tousif, S. et al. T cells from programmed death?1 deficient mice respond poorly to Mycobacterium tuberculosis infection. PLoS ONE 6, e19864 (2011).
- Lazar-Molnar, E. et al. Programmed death?1 (PD?1)- deficient mice are extraordinarily sensitive to tuberculosis. Proc. Natl Acad. Sci. USA 107, 13402–13407 (2010).
- Jayaraman, P. et al. TIM3 mediates T cell exhaustion during Mycobacterium tuberculosis infection. PLoS Pathog. 12, e1005490 (2016).
- Sotgiu, G., Centis, R., D’Ambrosio, L. & Migliori, G. B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect Med. 5, a017822 (2015).
- Attanasio, J. & Wherry, E. J. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity 44, 1052–1068 (2016).
- Harding, F. A., McArthur, J. G., Gross, J. A., Raulet, D. H. & Allison, J. P. CD28?mediated signalling co?stimulates murine T cells and prevents induction of anergy in T?cell clones. Nature 356, 607–609 (1992).
- Krummel, M. F. & Allison, J. P. CD28 and CTLA?4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 (1995).
- A seminal study of CTLA4.
- Freeman, G. J. et al. Engagement of the PD?1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 192, 1027–1034 (2000).
- A seminal study of the PD1–PDL1 interaction.
- Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD?1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
- Chemnitz, J. M., Parry, R. V., Nichols, K. E., June, C. H. & Riley, J. L. SHP?1 and SHP?2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but onlyreceptor ligation prevents T cell activation. J. Immunol. 173, 945–954 (2004).
- Francisco, L. M. et al. PD?L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 206, 3015–3029 (2009).
- Kamphorst, A. O. et al. Rescue of exhausted CD8 T cells by PD?1?targeted therapies is CD28?dependent. Science 355, 1423–1427 (2017).
- An important study highlighting the balance between immune activation and immune suppression.
- Yamazaki, T. et al. Expression of programmed death 1 ligands by murine T cells and APC. J. Immunol. 169, 5538–5545 (2002).
- Keir, M. E. et al. Tissue expression of PD?L1 mediates peripheral T cell tolerance. J. Exp. Med. 203, 883–895 (2006).
- Brown, J. A. et al. Blockade of programmed death?1 ligands on dendritic cells enhances T cell activation and cytokine production. J. Immunol. 170, 1257–1266 (2003).
- Liang, S. C. et al. Regulation of PD?1, PD?L1, and PD?L2 expression during normal and autoimmune responses. Eur. J. Immunol. 33, 2706–2716 (2003).
- Balar, A. V. et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet 389, 67–76 (2017).
- Rittmeyer, A. et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389, 255–265 (2017).
- Homet Moreno, B., Mok, S., Comin-Anduix, B., Hu?Lieskovan, S. & Ribas, A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunol. 5, e1052212 (2016).
- Latchman, Y. et al. PD?L2 is a second ligand for PD?1 and inhibits T cell activation. Nat. Immunol. 2, 261–268 (2001).
- Zhang, Y. et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl Acad. Sci. USA 103, 11695–11700 (2006).
- Tseng, S. Y. et al. B7?DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J. Exp. Med. 193, 839–846 (2001).
- First study to highlight a protective role for PDL2.
- Shin, T. et al. Cooperative B7?1/2 (CD80/CD86) and B7?DC costimulation of CD4+ T cells independent of the PD?1 receptor. J. Exp. Med. 198, 31–38 (2003).
- Shin, T. et al. In vivo costimulatory role of B7?DC in tuning T. helper cell 1 and cytotoxic T. lymphocyte responses. J. Exp. Med. 201, 1531–1541 (2005).
- Michot, J. M. et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur. J. Cancer 54, 139–148 (2016).
- Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363, 711–723 (2010).
- Topalian, S. L. et al. Safety, activity, and immune correlates of anti?PD?1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
- Brahmer, J. R. et al. Safety and activity of anti?PD?L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
- Weber, J. S., Postow, M., Lao, C. D. & Schadendorf, D. Management of adverse events following treatment with anti-programmed death?1 agents. Oncologist 21, 1230–1240 (2016).
- Topalian, S. L. et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 32, 1020–1030 (2014).
- Barber, D. L. et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 (2006).
- Wherry, E. J. et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684 (2007).
- Youngblood, B. et al. Cutting edge: prolonged exposure to HIV reinforces a poised epigenetic program for PD?1 expression in virus-specific CD8 T cells. J. Immunol. 191, 540–544 (2013).
- Ahn, E. et al. Demethylation of the PD?1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. J. Virol. 90, 8934–8946 (2016).
- Pauken, K. E. et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD?1 blockade. Science 354, 1160–1165 (2016).
- Sen, D. R. et al. The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169 (2016).
- An important paper defining how T cell exhaustion is controlled at the epigenetic level.
- Zhu, C. et al. The Tim?3 ligand galectin?9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252 (2005).
- Zhou, Q. et al. Coexpression of Tim?3 and PD?1 identifies a CD8+ T?cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 (2011).
- Sakuishi, K. et al. Targeting Tim?3 and PD?1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med. 207, 2187–2194 (2010).
- Huang, C. T. et al. Role of LAG?3 in regulatory T cells. Immunity 21, 503–513 (2004).
- Woo, S. R. et al. Immune inhibitory molecules LAG?3 and PD?1 synergistically regulate T?cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
- Dougall, W. C., Kurtulus, S., Smyth, M. J. & Anderson, A. C. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 276, 112–120 (2017).
- Lozano, E., Dominguez-Villar, M., Kuchroo, V. & Hafler, D. A. The TIGIT/CD226 axis regulates human T cell function. J. Immunol. 188, 3869–3875 (2012).
- Yu, X. et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 10, 48–57 (2009).
- Blake, S. J., Dougall, W. C., Miles, J. J., Teng, M. W. & Smyth, M. J. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin. Cancer Res. 22, 5183–5188 (2016).
- Gonzalez, L. C. et al. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc. Natl Acad. Sci. USA 102, 1116–1121 (2005).
- Sedy, J. R. et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat. Immunol. 6, 90–98 (2005).
- Cai, G. & Freeman, G. J. The CD160, BTLA, LIGHT/ HVEM pathway: a bidirectional switch regulating T?cell activation. Immunol. Rev. 229, 244–258 (2009).
- Fourcade, J. et al. CD8+ T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD?1. Cancer Res. 72, 887–896 (2012).
- Ephrem, A. et al. Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur. J. Immunol. 43, 2421–2429 (2013).
- Shimizu, J., Yamazaki, S., Takahashi, T., Ishida, Y. & Sakaguchi, S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat. Immunol. 3, 135–142 (2002).
- Schaer, D. A., Cohen, A. D. & Wolchok, J. D. Anti-GITR antibodies — potential clinical applications for tumor immunotherapy. Curr. Opin. Investig. Drugs 11, 1378–1386 (2010).
- Mitsui, J. et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin. Cancer Res. 16, 2781–2791 (2010).
- Deng, J., Le Mercier, I., Kuta, A. & Noelle, R. J. A new VISTA on combination therapy for negative checkpoint regulator blockade. J. Immunother. Cancer 4, 86 (2016).
- Gao, J. et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med, 23, 551–555 (2017).
- Freeman, G. J., Wherry, E. J., Ahmed, R. & Sharpe, A. H. Reinvigorating exhausted HIV-specific T cells via PD?1?PD?1 ligand blockade. J. Exp. Med. 203, 2223–2227 (2006).
- Kaufmann, D. E. & Walker, B. D. PD?1 and CTLA?4 inhibitory cosignaling pathways in HIV infection and the potential for therapeutic intervention. J. Immunol. 182, 5891–5897 (2009).
- Golden-Mason, L. et al. Upregulation of PD?1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J. Virol. 81, 9249–9258 (2007).
- Nakamoto, N. et al. Functional restoration of HCV-specific CD8 T cells by PD?1 blockade is defined by PD?1 expression and compartmentalization. Gastroenterology 134, 1927–1937 (2008).
- Lukens, J. R., Cruise, M. W., Lassen, M. G. & Hahn, Y. S. Blockade of PD?1/B7?H1 interaction restores effector CD8+ T cell responses in a hepatitis C virus core murine model. J. Immunol. 180, 4875–4884 (2008)
- Urbani, S. et al. Restoration of HCV-specific T cell functions by PD?1/PD?L1 blockade in HCV infection: effect of viremia levels and antiviral treatment. J. Hepatol. 48, 548–558 (2008).
- Rutigliano, J. A. et al. Highly pathological influenza A virus infection is associated with augmented expression of PD?1 by functionally compromised virus-specific CD8+ T cells. J. Virol. 88, 1636–1651 (2014).
- Sharma, S. et al. T cell immunoglobulin and mucin protein?3 (Tim?3)/Galectin?9 interaction regulates influenza A virus-specific humoral and CD8 T?cell responses. Proc. Natl Acad. Sci. USA 108, 19001–19006 (2011).
- Xu, D. et al. A potential new pathway for PD?L1 costimulation of the CD8?T cell response to Listeria monocytogenes infection. PLoS ONE 8, e56539 (2013).
- Bhadra, R., Gigley, J. P., Weiss, L. M. & Khan, I. A. Control of Toxoplasma reactivation by rescue of dysfunctional CD8+ T?cell response via PD?1?PDL?1 blockade. Proc. Natl Acad. Sci. USA 108, 9196–9201 (2011).
- Mou, Z. et al. Parasite-derived arginase influences secondary anti-Leishmania immunity by regulating programmed cell death?1?mediated CD4+ T cell exhaustion. J. Immunol. 190, 3380–3389 (2013).
- Esch, K. J., Juelsgaard, R., Martinez, P. A., Jones, D. E. & Petersen, C. A. Programmed death 1?mediated T cell exhaustion during visceral leishmaniasis impairs phagocyte function. J. Immunol. 191, 5542–5550 (2013).
- Joshi, T., Rodriguez, S., Perovic, V., Cockburn, I. A. & Stager, S. B7?H1 blockade increases survival of dysfunctional CD8+ T cells and confers protection against Leishmania donovani infections. PLoS Pathog. 5, e1000431 (2009).
Материал подготовлен НОИМТОиР: клинический ординатор Бриш Н.А.